Alexander Graham Bell, Father of Measuring Carbon Dioxide Accurately

There is, quite naturally, a confusion about how modern (post Eisenhower) measurements of CO2 have been made (there are changes underway). Even, for example, Spencer Weart, in his Discovery of Global Warming, gets it wrong
Keeling wanted to buy a new type of gas detector (namely, infrared spectrophotometers) that penned a precise and continuous record on a strip chart.Eli was reminded of this issue in a comment by one Izen over at Confused* Judy's place. (What, you doubt that Judy is Confused, Judith Curry, former chair and professor of atmospheric sciences over at Georgia Tech. Well, go read what Brian wrote last night first, and then what Confused Judy wrote yesterday about 2014 being the hottest year on record.) Izen:
I have no doubt that chemical and physical methods used in the past could be refined to a high accuracy. The technology of laser spectroscopic measurement was a more recent development than atom splitting. That provided Keeling with a better method.But the importance of maintaining that accuracy and maintaining control over the conditions, time position, that the measurements were made is Keeling’s contribution to the science.Now anybunny familiar with the spectrophotometers of the time, IR, Vis, UV (Beckman DU folks) would have some doubts about what kind of accuracy even one so obsessed as Charles Keeling could achieve.
To understand what and why start with a working definition that spectrometer disperses (or shuffle in the case of FT spectrometer) light of different frequencies so that the response of the sample can be measured as a function of frequency. As to the measurement, there are in principle two types of measurements that can be made. The most common one is absorption, where the intensity of the light at different frequencies/wavelengths, with and without the sample are measured. The second is excitation, where the response of the sample to the light is measured, typically by fluorescence or ionization, but also by the noise it makes when excited. Bunnies knew all about this from the year dot, but the photo-acoustic effect was first described by Alexander Graham Bell.
The advantage of an absorption measurement is that it is absolute. You only need to measure the relative amount of light with and without the sample and use the Beer Lambert law

where A is the absorbance and I and Io the intensities with and without the sample. The absorbance has a simple relationship with the cross-section σ, the length of the sample l through which the light passes (use your ruler) and the concentration of the interesting stuff N (and sometimes interferences in the sample)
A = σ l N
The difficulty of using absorbance for accurate and precise measurements of small concentrations is that if the difference between I and Io is small the absorbance is small and you would need to measure the light intensity to a precision and accuracy much higher than the electronics of yesteryear would allow and even today would be tough.**
Excitation spectroscopies are more sensitive, because in the absence of whatever absorbs the light the baseline is zero. There is a cost. Of course there is a cost, what do you think that there is free lunch at the hutch? The cost is that you need a calibrated sample whose concentration is accurately known so you can compare with. Although David Keeling had developed chemical methods and skill to measure CO2 in atmospheric samples accurately, each measurement was painstaking and for the kind of measurements that were needed in the pilot Mauna Loa program something better was needed.
Which is where Alexander Graham Bell, and a new instrument developed by V.N. Smith for IR measurements of various gases comes in. Jones realized that if what you want is to measure the effect of a gas on the intensity of light passing through a sample, you did not have to disperse the light and measure the effect at a single frequency, but rather you could compare the intensity passing through cells with and without the absorbing molecule
In general, however, I is a very small fraction of Io, so that a detector which is responsive to all wavelengths will be irradiated by a large amount of energy Io in the absence of X in the absorption cell, and by only a slightly smaller amount of energy I in the presence of X therein.
This difficulty can be overcome by irradiating two detectors, one through an absorption cell. containing X, and the other through an empty cell, or a cell containing a non-absorbing gas. The difference in the amount of energy received by the two detectors will be I, and a more sensitive indicating or recording instrument can be applied to the output from the two detecting elements. Although the calibration in this case will vary with the total energy Io, since the absorbed energy ex is a fraction thereof, this additional difficulty can be in turn overcome by using the null principle, that is, by stopping down the energy passing through the empty cell until the energy difference in the two detectors is zero, and then calibrating the action of the optical wedge used for this purpose in terms of concentration of the component X.This is called the Non-Dispersive IR method, often written as NDIR and is the basis of a whole raft of modern NDIR meters for monitoring CO2 and other gases in many application including medical agriculture and such. The characteristic of such meters is that they use a thin film filter to restrict the wavelength range of the light that reached the detector or they are used in a situation where there is only a single absorber.

Given the state of the art of dielectric IR filters in the 1950s, this would not be useful for the kinds of measurements that Keeling and Revelle wanted to make.
Enter Alexander Graham Bell's photo-acoustic detector. Photo-acoustic detectors can be exceedingly sensitive. As shown below they consist of a light source, a chopper and a microphone.
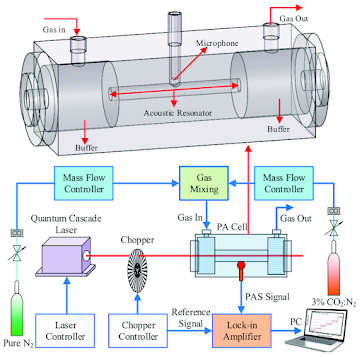
The first is a precise and accurately known calibration sample, a gas whose concentration is exactly (or as exactly as possible known). The current state of the art is described by NOAA where Peter Tans maintains the WMO international standard. IEHO, the lack of good calibration standards to test their results was a major failure of most of the pre-Mauna Loa CO2 measurement series. NOAA, relatively recently took possession of the standard cylinders from the Keeling labs at Scipps (1995). A calibration standard is a necessary bullshit test of any measurement. Still, anyrabbit who has ever made up calibration mixtures, especially at low concentrations knows that this is not bunny play. There are any numbers of materials issues, worrying about absorption and reaction on surfaces, issues associated with ensuring that the mixture is homogeneously mixed and don't talk about the issues with pumps.and any moving part in the system. Maintaining a sample over long periods is a horror which requires constant rechecking and not a little bit of hard experience.
The second, not so obvious, is the flow system that brings the sample and calibration gases into the cell. Again, materials are a major issue to build a system that samples from where you want the samples to come from, monitoring of meterorology, etc. Since measuring between the sample and the calibration gas are alternated (see this description of the NOAA SOP) if the measurement is to be automated this has to be done with electro (mechanical) valves. Design of the flow and sampling system is absolutely crucial
The third is REALLY obvious, you have to freeze out the water from the flowing sample gas.
* as in the George Bush sense
** as a side note trying to measure very high absorbances requires measuring a very small signal, down in the noise as it were. On commercial instruments ( which use base 10 logs to report absorbance) unless you have paid a lot for a special don't believe an absorbance base 10 above 2. Cut the concentration or make the length longer.



13 comments:
thanks, some types "this additional di(ff)iculty can be in turn overcome by using the null principle, that is, by stopping down the energy passing through the empty cell until the energy di(ii->ff)erence" I think I saw a 3rd one too, but missed it when reading again.
Thanks for the heads up fixed the two.
Eli
But as pointed out first by Tyndall and later by Einstein, gases do not just absorb, they also emit. Therefore what is measured is net absorption, and not total absorption.
The climate models equate total absortion with emission, and get a heating effect of zero [Goody & Yung, Equ. (2.80),p. 40.] The Einstein coefficient of absorption equals the the sum of the coefficients of emission, but only in thermodynamic equilibrium which is not true for the Earth's atmosphere. It is not surprising that the models and the data do not agree.
Well, said Eli, a) that's why you compare a calibrated sample against the measurement and b) please explain how IR spectrometers can possibly work if you are right.
(Hint: The intensity of the IR entering the cell from the light source is much higher than the emission rate which is limited by the temperature of the gas. This is something that trips up a lot of analytical chemists of Eli's acquaintance wrt to what goes on in the atmosphere so don't feel bad)
I am off topic, since I am talking about the greenhouse effect, not measuring CO2 concentration. However, it is related and what I am arguing is something that has been bothering me recently.
"The intensity of the IR entering the cell from the light source is much higher than the emission rate which is limited by the temperature of the gas." Yes, and you get an absorption spectrum.
But the spectrum is formed from two components; the unabsorbed IR that entered the cell and the radiation emitted by the gas due to induced and spontaneous emissions.
It seems to me that the emission componenet is being ignored when Einstein's B01 coefficient is being calculated for the HITRAN database etc.
HITRAN lists the Einstein A coefficient from which you can get the Bs. Also the air broadening, the self broadening, the temperature dependence of the line width and the pressure shift.
It's all there.
Apart from inspiring nostalgia for my long lost Beckman DU, this provides just cause for pimping my paean to Tyndall's great rock salt steampunk experiment
Yes, I see now. The A coefficent is not calculated from the B coefficients. It calculated from the intensity here. The B coefficients can then be calculated from the A coefficient.
Mostly for IR, for UV/Vis you can calculate A from radiative lifetime and then get B from it. You have one, then you have the other
I am surprised that you can calculate A, 1/(radiative lifetime), for CO2 vibrations since A increases with the cube of the frequency. At low frequencies, such as the far infrared and microwave, stimulated emissions dominate, because small values of A will result in large radiative livetimes, and the excited states will be relaxed by collisions rather than spontaneously.
But there is a problem. You do not consider stimulated emissions in your calculations.
Only needed in the upper Martian atmosphere such as it is. (also see Venus) Otherwise no need.
The fact that stimulated emission happen in the Martian and Venusian atmosphere does not prevent it happening in the Earth's atmosphere. In fact it makes it more likely since why should the Earth be different.
And stimulated emissions do not require a population inversion; that is only necessary for lasing.
If you look at the spectrum at the TOA, I think you will find it is produced by stimulated emissions, not absorption.
Post a Comment